A Computational Study of Ivermectin and Doxycycline Combination Drug Against SARS-CoV-2 Infection
- ivermectinforcovid

- Sep 24, 2021
- 8 min read
Abstract
In the present study, we have described how by using molecular docking and molecular dynamics (MD) simulation studies the combination drug of ivermectin and doxycycline can be used as a potential inhibitor for Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) virus. Instead of the unavailability of a specific cure for coronavirus disease of 2019 (COVID-19) till now various possibilities for individual and combination drugs have been explored by the medical practitioners/scientists for the remedial purpose of CoV-2 infections. 3C-like protease (3CLpro) is the main protease of the SARS-CoV-2 virus which plays an essential role in mediating viral replication in the human body. 3CLpro protein can serve as an attractive drug target. In this work, we have studied drug: 3CLpro interactions by in-silico molecular docking and MD simulation approaches. Common and easily available antiviral drugs ivermectin, doxycycline, and their combination can regulate 3CLpro protein's function due to its easy inhibition.
Introduction:
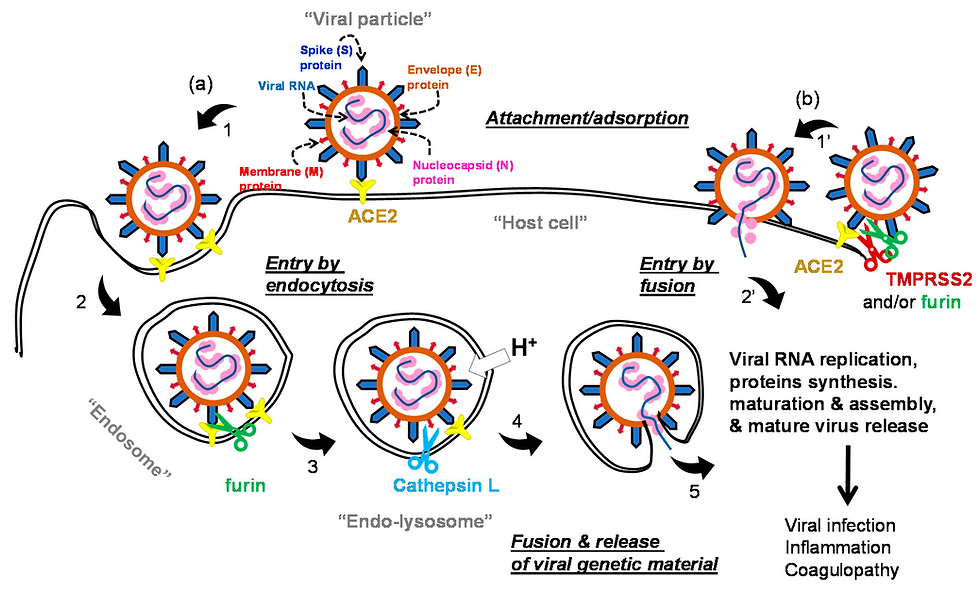
In the year 2020, the COVID-19 disease has spread globally and it has become an ongoing pandemic. As reported by the World Health Organization (WHO), due to this pandemic disease, more than 35,659,007 numbers of active patients with 1,044,269 people have already died till 10 October 2020 (https://covid19.who.int/). WHO declared the COVID-19 as a global health emergency. This disease is caused by a member of the coronavirus family [1]. Coronavirus was first found in 1930 in domestic poultry [2]. After that, they were identified as causing several diseases in humans such as; respiratory illness, neurological, liver diseases, etc. [3]. Till now seven categories of this virus were identified. Among the seven categories of coronavirus, four cause only common cold with mild symptoms and in very rare cases pneumonia, respiratory infections in infants and older people [4]. The other three categories are Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) [5], Middle East Respiratory Syndrome Coronavirus (MERS-CoV) [6] and lastly, the new one known as SARS-CoV-2 [7] identified in 2003, 2012, and 2019 respectively. The international committee on taxonomy of viruses declared this new novel coronavirus as SARS-CoV-2 [8]. The SARS-CoV-2 is a single-stranded RNA virus and belongs to the Coronaviridae family having genome sequences of 79.5% sequence matching [9,10]. This shows that bats may be the carrier of this virus. The uniqueness of this virus is the presence of spike glycoproteins on its surface which gives a crown-like appearance to the virus structure. The crown-like spike protein surface of this virus can be easily visible with the help of electron microscopes. These spike proteins are a very significant part of the SARS-CoV-2 [11] virus as they can easily interact with the human proteins which coat the inside of the nose and the cells of the lungs. The interaction of spike protein and human protein causes a change in the spike protein of CoV-2 shape and causes the human receptor cell to swallow up the virus. Through the receptor-binding domain (RBD), the glycoproteins of the viruses start binding and entering the host cells. The key receptor for SARS-CoV-2 in humans is an angiotensin-converting enzyme 2 (ACE2) [12]. After entering the host cell, different human protease like airway trypsin-like protease (HAT),
Materials and Methods:
2.1. Protein structure preparation Coronavirus possesses several polyproteins (structural and nonstructural). Among them, 3CLpro is a key CoV enzyme that plays an important role in mediating viral replication and transcription with the help of its glycoprotein. To rapidly discover the targeted drugs for clinical use, researchers focused on identifying drug leads that target the 3CLpro protein of SARS-CoV-2 as it plays an important role in viral replication and transcription. In the present work, we have used one of the 3CLproproteases of CoV-2 virus in a complex with an inhibitor N3 (PDB ID: 6LU7) [46,47] as the target protein. 6LU7 is a promising target for designing COVID-19 drugs. We have chosen 6LU7 for checking the inhibiting and binding properties of it with the ivermectin and doxycycline drugs. The structure of SARS-CoV-2 protease (PDB ID: 6LU7) was used as a receptor and retrieved from Protein Data Bank (http://www.rcsb.org/) [48,49] and are shown in Figure 1 (a). We have removed water and hydrogen from it. All the existing properties of the drugs are described in Table 1. For the preparation of protein, we have used Auto Dock and MG Tools of AutoDockVina software [50]. At first existing lead components, water molecules and ions have been removed from it. Later the process of cleaning has been done. We have calculated the Gasteiger charges of protein structures and after that polar hydrogen has been introduced. Then the non-polar bonds were merged and rotatable bonds were defined. Finally, by using Discovery Studio 2020 [51] the intrinsic ligands were detached from the protein molecules and the final protein molecule was saved in the PDB format (Figure 2 a). Ligand drug molecules preparations Structures of the drug molecules were downloaded from Drug Bank in PDB format. Then this structure was fully optimized by using the Gaussian 09 program [52]. We have used the optimized structure for docking analysis as they provide better results than unoptimized ones. The geometric optimization of all drug compounds was carried out using Hartree-Fock (HF) and STO 3G basis set. Gauss View 5 molecular visualization program was used for visualizing the optimized structure [53]. ADME-T properties of molecules were identified using the Organic chemistry portal (http://www.organicchemistry.org/prog), a web-based application for predicting in-silicoADME-T property. Protein-ligand interactive visualization and analysis were carried out in AutoDock 4.2 software on Windows 7 (64-bit). For the present work, we have selected two potential ligand drugs: ivermectin (C48H72O14) and doxycycline (C22H24N2O8). Detail structures of these molecules were downloaded from Drug Bank in PDB format (Figure 1 and Table 1). Different chemical, physical, drug-likeness, and pharmacokinetics properties obtained from SWISS ADME are shown in Table 1. Both the proposed drug molecules have a molecular weight less than 875 gm/mol and topological polar surface area (TPSA) values less than 180 Å. 2 (Table 1). All drug molecules have H-bond donors ≥6, H-bond acceptor ≥14, and have a low synthetic accessibility count, this suggests that they can be synthesized easily. Though these drugs violate some 8 drug-likeness properties, still the availability of these drugs in the drug industry motivates us to consider these as potential inhibitors. The ligand file in pdbqt format is needed for molecular docking study with AutoDock Tools. AutoDock Tools 1.5.6 [54] have been used to save ligands in pdbqt format.




Comments